Las enfermedades minoritarias (también llamadas raras o poco frecuentes) se definen como aquellas que afectan a 1 o menos de cada 2.000 personas. En conjunto existen cerca de 10.000 tipos de patologías consideradas minoritarias, muchas sin tratamiento. La investigación es la única estrategia para desarrollar terapias por estas personas.
Las distrofias hereditarias de la retina (DHR) son un grupo heterogéneo de enfermedades raras oculares causadas por la mutación de un solo gen (monogénicas o Mendelianas). Se conocen más de 300 genes causantes de las CHR
A continuación encontrará más información sobre cada una de estas:
La enfermedad de Stargardt (OMIM 248200) es una de las distrofias hereditarias de retina macular más frecuente. La mayoría de los casos se heredan de forma autosómica recesiva. La disminución de visión que comporta esta enfermedad afecta habitualmente a personas jóvenes, con una incidencia sobre todo en adolescentes y adultos jóvenes menores de 20 años (1:10.000). La enfermedad de Stargardt y el fundus flavimaculatus son dos presentaciones clínicas de la misma enfermedad. A nivel histológico, se produce un cúmulo de material tipo lipofuscina en las células del epitelio pigmentario de la retina por la mutación del gen ABCA4. Esta mutación se transmite cuando ambos progenitores también poseen la malformación de ese gen.
La enfermedad de Stargardt provoca una visión desenfocada y sin nitidez, que dificulta reconocer rostros y formas, y leer tanto de cerca como de lejos y que al final induce a confundir colores de matices cercanos (por ejemplo, negro y azul marino). También causa dificultad para adaptarse a la penumbra. Aunque no provoca ceguera absoluta, las personas que la padecen pueden perder agudeza visual hasta llegar a la ceguera legal.
Actualmente, no existe ningún tratamiento para esta patología, pero la investigación en este campo sigue siendo muy activa. Existen centros en Europa y Estados Unidos que están participando en distintos ensayos clínicos con el objetivo de encontrar una terapia efectiva para los pacientes afectados de Stargardt. Estas investigaciones incluyen tanto un acercamiento farmacológico como terapias génicas y con células madre.
Los principales tratamientos farmacológicos se dividen en dos grandes grupos: los moduladores del ciclo visual y los inhibidores del complemento. El Institut de la Màcula, en estrecha colaboración con la Barcelona Macula Foundation (BMF), es el único centro en España que está participando en el ensayo clínico que tiene por objetivo probar la eficacia y seguridad de Zimura® ( IVERIC bio, estudio en fase 2b, multicéntrico: OPH2005) en pacientes con Stargardt.
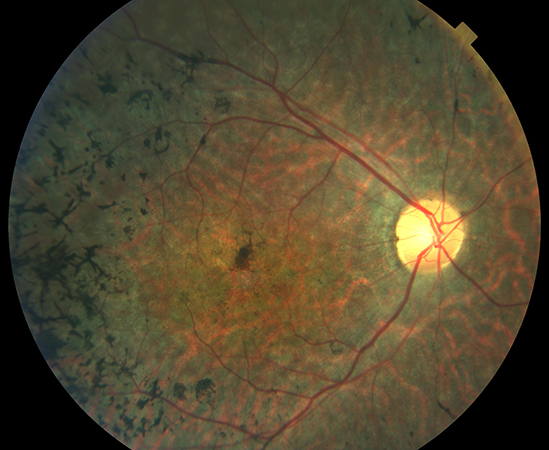
La retinosis pigmentaria (RP) es una enfermedad ocular de carácter degenerativo y hereditario que provoca una disminución grave de la capacidad visual y que, en muchos casos, termina en ceguera. Con una afectación de 1:4.000, en España el número de afectados supera los 15.000 y se estima que 60.000 personas son portadores de los genes defectuosos y responsables de la enfermedad y, por tanto, posibles transmisores de ésta. La aparición de la patología afecta generalmente a personas jóvenes y los primeros síntomas son la dificultad de adaptación a la oscuridad y una pérdida progresiva del campo visual.
La retinosis pigmentaria describe un grupo de enfermedades hereditarias de la retina que se caracterizan por una pérdida progresiva de los fotorreceptores (apoptosis), sobre todo los de tipo bastón, y del epitelio pigmentario de la retina, debido a mutaciones de proteínas y enzimas específicas de la misma. El componente hereditario está presente en la mitad de los casos de retinosis pigmentaria y el pronóstico, así como su progresión, puede estar relacionado con esa herencia.
Actualmente no se dispone de ningún tratamiento eficaz para combatir la retinosis pigmentaria. La identificación del gen responsable de la enfermedad es un aspecto fundamental para el manejo clínico del paciente con RP por la terapia génica. Esto permite no sólo la confirmación del diagnóstico, sino también establecer la correlación genotipo/fenotipo, ofrecer un pronóstico más acertado y, en el futuro próximo, indicar un tipo de tratamiento u otro.
En la actualidad, investigadores de la Barcelona Macula Foundation han iniciado el proyecto DRUG4SIGHT financiado por La Caixa con el IBEC (Institut de Bioenginyeria de Catalunya) y otras entidades de ámbito estatal para desarrollar nuevos fármacos en pacientes con degeneraciones de la retina. El paradigma de este proyecto es completamente distinto de las demás aproximaciones terapéuticas. Aquí se pretende descubrir y caracterizar una serie de fármacos que puedan estimular proteínas todavía presentes en la retina degenerada y hacer actuar a las células remanentes no degeneradas como fotorreceptores, células sensibles a la luz. Los resultados preliminares son prometedores, pero habrá que confirmarlos en modelos animales con un sistema visual más parecido al ser humano.
Coroideremia
La coroideremia (OMIM 300390) es una enfermedad recesiva causada por mutaciones en el gen CHM, localizado en el cromosoma Xq21.2. La mutación provoca la carencia de una proteína, el Rab escort protein 1 (REP1), que se cree que está involucrada en el tráfico intracelular y/o la función de los bastones. Afecta típicamente a hombres (1:50.000) y las mujeres son portadoras.
La enfermedad provoca una pérdida de la coriocapilar (la capa de capilares más internos de la coroides), el epitelio pigmentario de la retina y los fotorreceptores (fundamentalmente los bastones). Se inicia en la media periferia y progresa de forma centrípeta hacia la mácula, con unos márgenes entre retina afectada y sana característicamente bien definidos. Los síntomas se inician habitualmente en la juventud. Los pacientes notan una pérdida del campo visual y empeoramiento de la visión nocturna que progresan hacia la disminución de la visión central de forma similar a la que se observa en pacientes con retinosis pigmentaria.
Aunque no tiene tratamiento, la coroideremia es una de las enfermedades hereditarias en las que existen más estudios intervencionales registrados (15 en www.clinicaltrials.gov, accedido el 3 de diciembre de 2019). Debido a que es una enfermedad monogénica, causada por alteraciones en un solo gen, el tratamiento mediante terapia génica en el que se reemplaza al gen afectado por uno que funciona con normalidad se está probando con resultados prometedores. Hay ensayos en muchos países, incluyendo un ensayo clínico en fase III en EE.UU. y, Europa, el estudio STAR.
Amaurosis congénita de Leber
La amaurosis congénita de Leber (ACL) es una enfermedad habitualmente autosómica recesiva presente en el momento de nacer o poco después. Se han descrito muchas mutaciones en genes distintos como responsables de su desarrollo. Se estima que tiene una prevalencia de 1:40.000 y puede asociarse a otras alteraciones sistémicas (retraso del desarrollo, disfunción renal, etc.).
La ACL se caracteriza por una degeneración retiniana, en la que un fondo de ojo aparentemente normal progresa hacia alteraciones pigmentarias generalizadas, atenuación vascular, atrofia retiniana y del nervio óptico. La visión está severamente afectada, y los bebés pueden manifestar nistagmus (movimiento oscilatorio de los ojos), fotofobia (sensibilidad anormal a la luz) o estrabismo (mal alineamiento de los ejes visuales).
En la mayoría de los casos no existe tratamiento, aunque se pueden dirigir algunas condiciones asociadas a la enfermedad (alta hipermetropía, cataratas, etc.) Sin embargo, un 10% de los casos de ACL se deben a mutaciones en el gen RPE65 (ACL tipo 2, OMIM 180069), y en estos casos se puede utilizar Luxturna® (voretigene neparvovec, de Spark Therapeutics), una terapia génica basada en el reemplazo del gen defectuoso por un gen de funcional que mostró buenos resultados visuales. En enero de 2018 obtuvo la aprobación por parte de la FDA (Food and Drug Administration) americana, y en abril de 2019 por la AEMPS (Agencia Española de Medicamentos y Productos Sanitarios). La aprobación de este fármaco ha sido un hito, al ser la primera terapia génica en oftalmología aprobada por una agencia reguladora.
Retinosquisis ligada a X
La retinosquisis ligada a X (OMIM 312700) es un trastorno ocular bilateral que aparece durante la infancia, causado por mutaciones en el gen RS1 localizado en el cromosoma Xp22.13. Como su nombre indica, se transmite ligada al cromosoma X. Afecta sólo a los hombres (1/5.000-1/25.000) pero las mujeres son portadoras y tienen un 50% de posibilidades de transmitir la mutación a sus hijos.
El gen mutado en la retinosquisis ligada a X (RS1) codifica por la retinosquisina, una proteína adhesiva que participa en la integridad estructural y funcional de la retina. Así, el fondo de ojo se caracteriza por la presencia de quistes en la zona foveal de la retina y estriaciones radiales. Los afectados presentan una disminución de la visión central y dificultades en la lectura. La pérdida se estabiliza durante la juventud para volver a disminuir hacia los 50-60 años.
A pesar de no haber tratamiento aprobado para la enfermedad, hoy en día existen activos dos ensayos clínicos (www.clinicaltrials.gov consultado el 3 de diciembre de 2019) para evaluar la seguridad, la tolerabilidad y la eficacia de un tratamiento con terapia génica.
Algunas de las complicaciones secundarias a esta enfermedad son el desprendimiento de retina (5-22%) y la hemorragia vítrea (4-40%). Por ello, a pesar de no haber tratamiento actualmente, se recomienda la revisión periódica para prevenir o tratar de forma inmediata cualquier complicación.
Atrofia Gyrata
La atrofia Gyrata (OMIM 258870) es una enfermedad rara causada por mutaciones en el gen que codifica la ornitina aminotransferasa OAT (10q26). La deficiente actividad de esta enzima produce hiperornitinemia, pero se desconoce todavía el mecanismo por el que desemboca en atrofia coriorretiniana. Es de herencia autosómica recesiva y su prevalencia es desconocida (se estima 1/50.000). La edad de aparición es variable (1 mes-44 años).
Los primeros síntomas son ceguera nocturna y reducción del campo visual producidos por múltiples áreas circulares de atrofia coriorretiniana en la periferia. Con el paso de los años, las áreas atróficas aumentan de tamaño convergiendo hacia la mácula y resultando en una pérdida de la visión central. Los pacientes pueden presentar asociada una miopía con marcado astigmatismo, catarata subcapsular posterior de inicio temprano y edema macular quístico.
Se ha descrito en varios estudios que una dieta restringida en arginina (precursora de la ornitina) o una dieta baja en proteínas puede reducir el nivel sérico de ornitina y retrasar así la progresión de la atrofia coriorretiniana y de la pérdida visual. Por este motivo es especialmente importante tener presente esta enfermedad, ya que la intervención dietética puede ralentizar su progresión. Aunque se han realizado algunos ensayos clínicos con terapia génica, actualmente no existe ninguno activo (www.clinicaltrials.gov consultado el 3 de diciembre de 2019).
Fundus Albipuntactus
El fundus albipuntactus (OMIM: 136880) es una de las distrofias hereditarias de la retina que debuta en la infancia. Esta patología es hereditaria, puede ser autosómica dominante o recesiva y su prevalencia es desconocida. La forma autosómica dominante se produce por una mutación en el gen PRPH2 y la forma recesiva se asocia a una mutación en el gen RDH5.
El gen RDH5 está involucrado en el ciclo visual. Este gen es el encargado de proporcionar las instrucciones a la enzima 11-cis retinol deshidrogenasa 5, que juega un papel importante en la codificación de la luz en señales eléctricas. Por eso, las mutaciones de éste provocan un déficit en la función que realiza la enzima 11-cis retinol deshidrogenasa 5, alterando el ciclo visual y, en consecuencia, la visión, sobre todo en condiciones de baja iluminación.
A nivel ocular, el fundo albipuntactus se caracteriza por la aparición de numerosas lesiones retinianas pequeñas redondeadas y de color blanco-amarillento que se distribuyen sobre todo en media periferia, sin alteración macular.
Los pacientes afectados con esta enfermedad presentan una ceguera nocturna no progresiva (con tiempos prolongados de adaptación de luz a oscuridad), por lo que es importante diferenciarlos de otras entidades progresivas con síntomas similares, como la retinosis pigmentaria. Su visión en condiciones de iluminación normales está conservada.
En la actualidad, en Estados Unidos se está llevando a cabo un estudio que se encarga de registrar en una base de datos a pacientes con enfermedades hereditarias degenerativas de la retina. La creación de estas bases de datos permitirá un mayor conocimiento de la enfermedad y su historia natural, así como aportar datos sobre su prevalencia.
La distrofia macular viteliforme de Best
La distrofia macular viteliforme de Best (BVMD) (OMIM 153700) es una enfermedad que debuta en la edad infantil y en la adolescencia. Esta patología presenta una prevalencia de 1 a 9 casos de cada 100.000, afecta más a hombres que a mujeres (3:1) y es hereditaria autosómica dominante.
En la mayoría de los casos, la BVMD es causada por una mutación en el gen BEST1 (localizado en el cromosoma 11q12). Este gen es el encargado de codificar la bestrofina-1, un canal de cloro que se expresa en el epitelio pigmentario de la retina, por lo que una alteración en esta proteína provoca la acumulación de lipofuscina a consecuencia de un intercambio iónico anómalo.
La BVMD se caracteriza por una lesión amarillenta en la mácula en forma de yema de huevo, debido a la acumulación anómala de lipofuscina entre los fotorreceptores y el epitelio pigmentario de la retina. En estadios más avanzados, se produce una atrofia del epitelio pigmentario de la retina. Los pacientes que padecen esta enfermedad presentan, por norma general, una disminución de la agudeza visual central, metamorfopsias, protanopía y disminución del índice de Arden en el electrooculograma. Por otra parte, la visión periférica y la adaptación a la oscuridad son normales.
Con el tiempo, la BVMD puede evolucionar hacia una atrofia geográfica y una de las complicaciones asociadas es la aparición de una membrana neovascular subfoveal coroidal, aunque no suele presentarse en niños.
Actualmente, los tratamientos que se llevan a cabo en pacientes con BVMD son para tratar las posibles complicaciones derivadas de la enfermedad, como es el uso de anti-VEFG en caso de que aparezca una membrana neovascular subfoveal coroidal. Los estudios realizados en los últimos años han permitido identificar los distintos patrones de autofluorescencia presentes en esta patología. Por ello, es importante realizar un seguimiento de la enfermedad a largo plazo, ya que las modificaciones clínicas en la autofluorescencia permitirán un mayor conocimiento de la misma y su pronóstico en función del patrón. Por otra parte, en Estados Unidos se están realizando diferentes estudios que incluyen la terapia génica y con células madre.
Síndrome de Usher
El síndrome de Usher (SU) es una enfermedad hereditaria autosómica recesiva que se caracteriza por la asociación de la sordera neurosensorial (generalmente congénita) con la retinosis pigmentaria (RP) y la pérdida progresiva de visión. El inicio generalmente ocurre durante la infancia y tiene una prevalencia de 1-9:100.000 (en Europa, de 3-4:100.000).
Se han identificado 3 tipos de SU basándose en las diferencias de la función auditiva y vestibular (la retinosis pigmentaria es común en los tres tipos): (a) tipo 1, en el que la pérdida de audición es congénita, profunda y no existe función vestibular; la retinosis pigmentaria no se manifiesta en el nacimiento y las mutaciones implican cinco genes (MYO7A, USH1C, CDH23, PCDH15, USH1G) y un locus (USH1E); (b) tipo 2, con una sordera congénita menos severa con función vestibular preservada, y mutaciones en los genes USH2A, GPR98 y DFNB31 y posible locus (15q); y (c) tipo 3, con inicio más tardío tanto de la sordera como de la retinosis pigmentaria, con mutaciones en CLRN1.
A pesar de las muchas investigaciones sobre el síndrome, actualmente no existe tratamiento. La investigación se centra en el desarrollo de terapias génicas mediante inyecciones subretinianas, implantes intraoculares de agentes neuroprotectores o los llamados “ojos biónicos”. Se debe tener en cuenta la necesidad de trabajar con un equipo multidisciplinar que también incluya otorrinolaringólogos, logopedas y psicólogos. Los implantes cocleares pueden ayudar a mantener un resto auditivo.
Distrofia de Conos
La distrofia de conos (OMIM 602093) es un grupo de enfermedades hereditarias retinales con un patrón autosómico recesivo o dominante. Se presenta generalmente en la infancia o en los primeros años de la vida adulta y su prevalencia es desconocida, mientras que en la distrofia conjunta de conos y bastones se estima que se encuentra alrededor de 1:40.000 personas.
Los síntomas característicos son la pérdida progresiva de agudeza visual, visión del color y fotofobia (sensibilidad anormal a la luz). En el electrorretinograma están afectados sólo los conos, con preservación de bastones. En algunos casos, en fases avanzadas pueden verse afectados los bastones, denominándose distrofia de conos y bastones; en estos casos el electrorretinograma muestra también afectación de los bastones. Oftalmoscópicamente, estas distrofias hereditarias de la retina se caracterizan por la presencia de depósitos de pigmentos en la región macular, que puede adquirir un aspecto de “ojo de buey”.
Tanto la distrofia de conos como la de conos y bastones se han asociado a mutaciones en el gen GUCA1A y en los cromosomas 6q, 17p y 19q. Otras enfermedades donde existe una afectación selectiva de los conos incluyen la acromatopsia o el síndrome de incremento de los conos S. Las alteraciones en la visión del color leves son entidades diferentes de la distrofia de conos.
Actualmente no existe un tratamiento que detenga la evolución de esta condición y restaure la visión, pero existen estrategias dirigidas a ayudar a los pacientes a atenuar el impacto social y psicológico de la pérdida visual. Las ayudas de baja visión (sistemas de aumento, filtros) pueden mejorar la calidad de vida de estos pacientes.
Bibliografía
Yannuzi, L. A. (2016) The retina atles. Elsevier
Agarwal, A (2011) Glass’ Atlas of macular diseases. Elsevier
Online Mendelian Inheritance in Man (2019). Disponible en http://www.omim.org.
Clinical Trials (2019). Disponible en http:///www.clinicaltrials.org
El portal sobre enfermedades raras y medicamentos huérfanos (2019). Disponible en http://www.orpha.net
Autors: Clara Abadías, Marc Biarnés, Miriam Garcia i Cristina Romero